1. Ferroelectricity: Pb-based and beyond
PbTiO3 and its solid solutions dominate the ferroelectric and piezoelectric industry. The exceptional polar and piezoelectric properties of these materials originate from the strong polar activity of the Pb2+ ion and the Pseudo Jahn-Teller effect of Ti4+ ion in its oxygen octahedral cage. But all is not well with Lead based compounds as Lead is toxic and needs to be eliminated. The idea is to replace lead by isovalent elements to get comparable or yet better ferroelectric properties. The closest isovalent cousin to Lead is Tin and is expected to show similar electronic properties. Preliminary first principles calculations on SnTiO3 show that Sn2+ ion exhibits even stronger polar activity. But several attempts to synthesize polar SnTiO3 were unsuccessful. One such attempt mostly yielded centrosymmetric (R3) structure with only traces of perovskite structure.
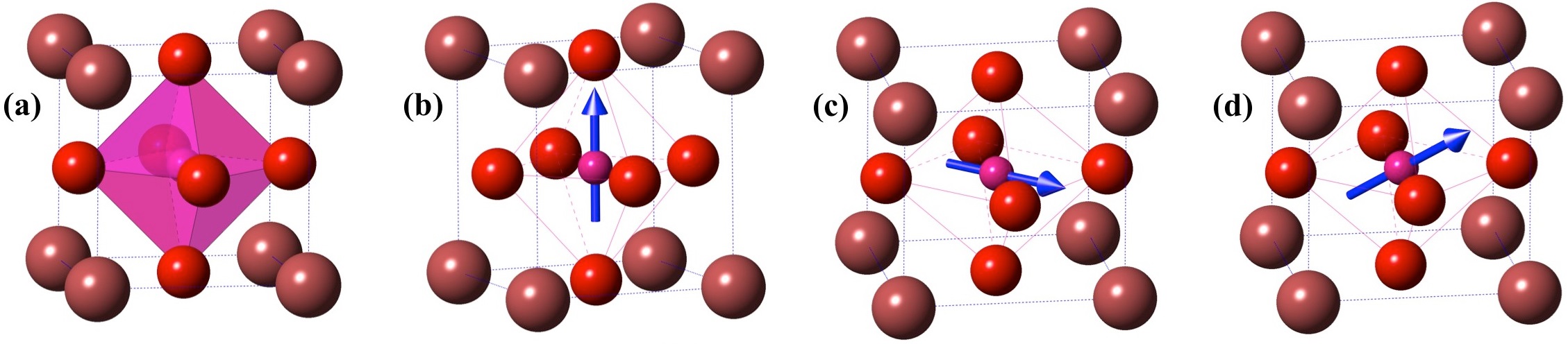
Fig.1: Crystal models of plausible polymorphs of ABO3 type compounds. (a) Pm3m (b) P4mm (c)Amm2 and (d) Cm. Atom A (Maroon) is located at the corner of the unitcell, B (Pink) at the body-centre and O (Red) at the face-centre. In non-polar Pm3m shown in (a), B-atom is at the geometric center of the octahedron. In other polar structures shown in (b), (c) and (d) B-atom is distorted in <0,0,c>, <a,a,0> and <a,a,c> direction as represented by blue arrows.
2. Energetic ordering of SnTiO3 polymorphs @ absolute zero
Structurally, the polar polymorphs can be obtained from the non-polar polymorph by distortions of A and B ions in specific directions as mentioned above. Depending on the external conditions including stress and chemical environment, these polymorphs energetically compete with each other. Under neutral conditions, both PbTiO3 and SnTiO3 assumes tetragonal P4mm structure. The plot below shows the energetic ordering of plausible polymorphs of SnTiO3 for a range of compressive and tensile pressures.

Fig-2: An investigation of stability of various SnTiO3 polymorphs done by calculating their optimized structures within the DFT/LDA. Plausible polymorphs include polar and nonpolar perovskites and trigonal R3c [lithium niobate (LN)] phases, all of which have corner-sharing TiO6 octahedra. Layered hexagonal phases are also feasible: R3 (ilmenite), P63/mmc, and P63mc structures, the former containing edge-sharing and the latter two face-sharing TiO6 octahedral networks.The connectivity of the octahedral network strongly affects the presence or absence of polar distortions in a given polymorph. The phases with corner-sharing octahedra are, in general, more stable than the hexagonal polymorphs. The polar perovskite (P4mm) phase has the lowest energy of all structures, and the two other low-energy phases are monoclinic perovskite Cm and LN-type R3c.The figure shows energy differences per formula unit for various SnTiO3 polymorphs with respect to the polar perovskite P4mm phase, which has the lowest energy. Energy variations with respect to slight phase volume change are also presented. Layered hexagonal phases have much higher energies, compared to the other phases, and their volume-energy curves are not shown here explicitly.
3. Sterical activity of Sn2+ and Pb2+ lone pairs:
| P4mm | Amm2 | Cm | Pm3m | |
| SnTiO3 | ||||
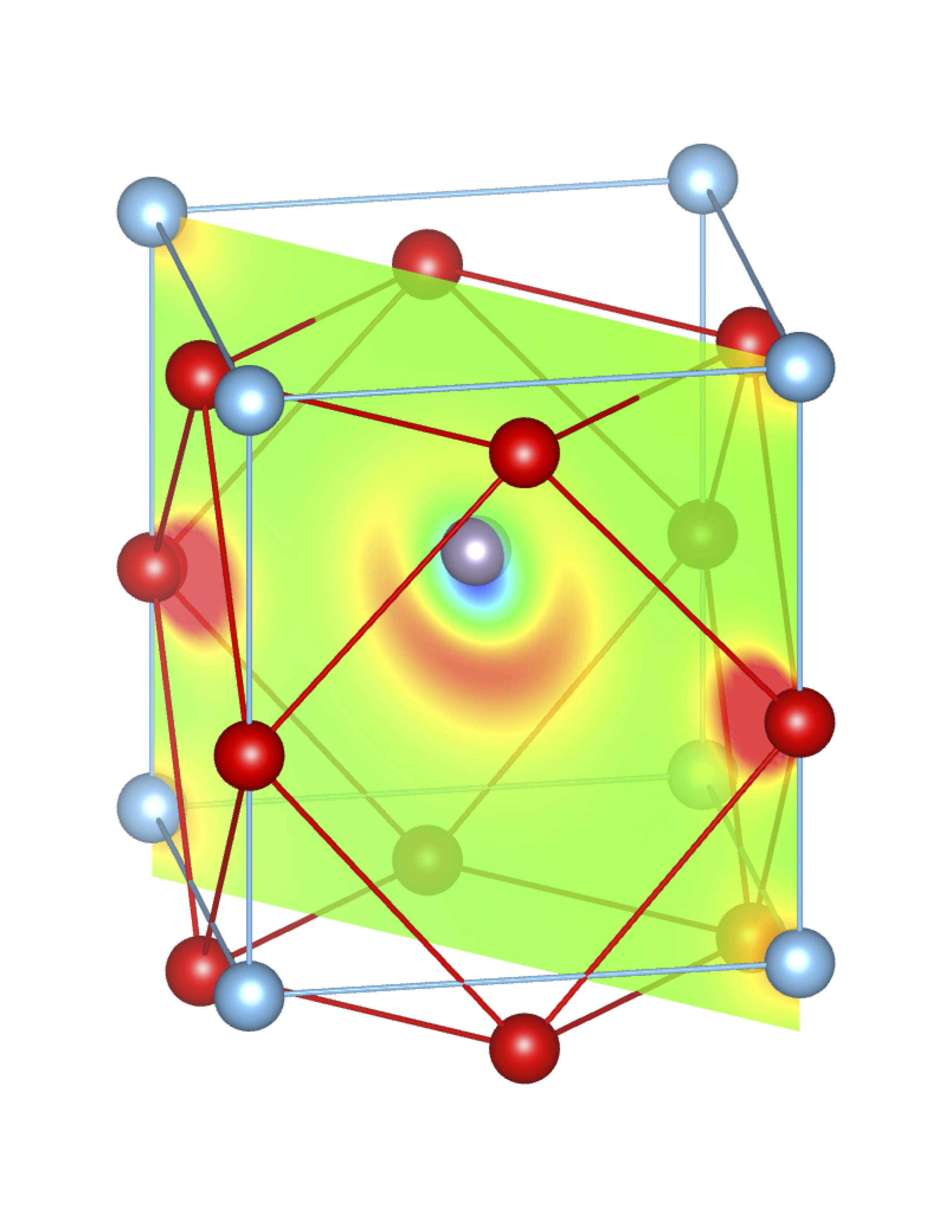 Total Charge (Q) = 0.58 e Total Charge (Q) = 0.58 e |
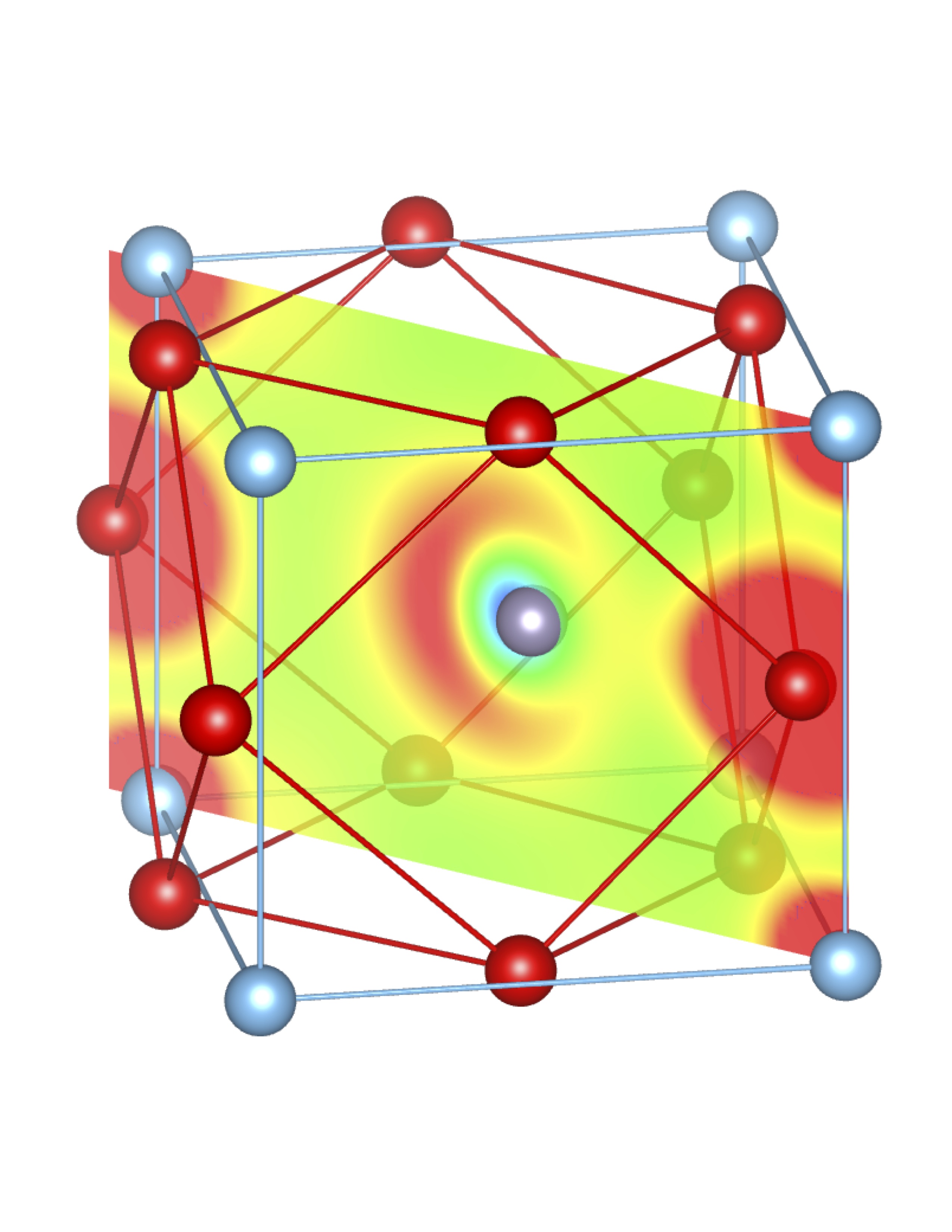 Total Charge (Q) = 0.74 e Total Charge (Q) = 0.74 e |
 Total Charge (Q) = 0.57 e Total Charge (Q) = 0.57 e |
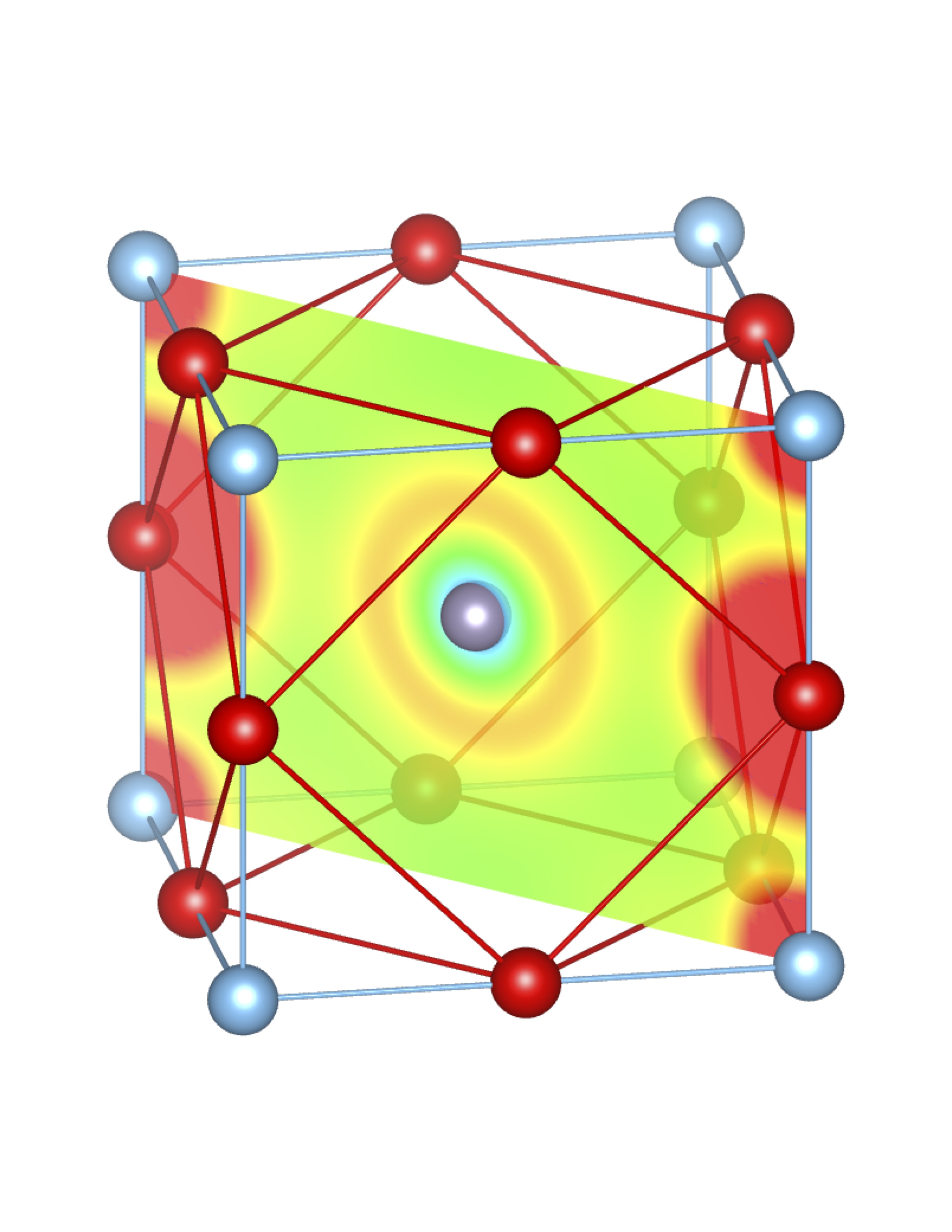 Total Charge (Q) = 0.47 e Total Charge (Q) = 0.47 e |
|
| PbTiO3 | ||||
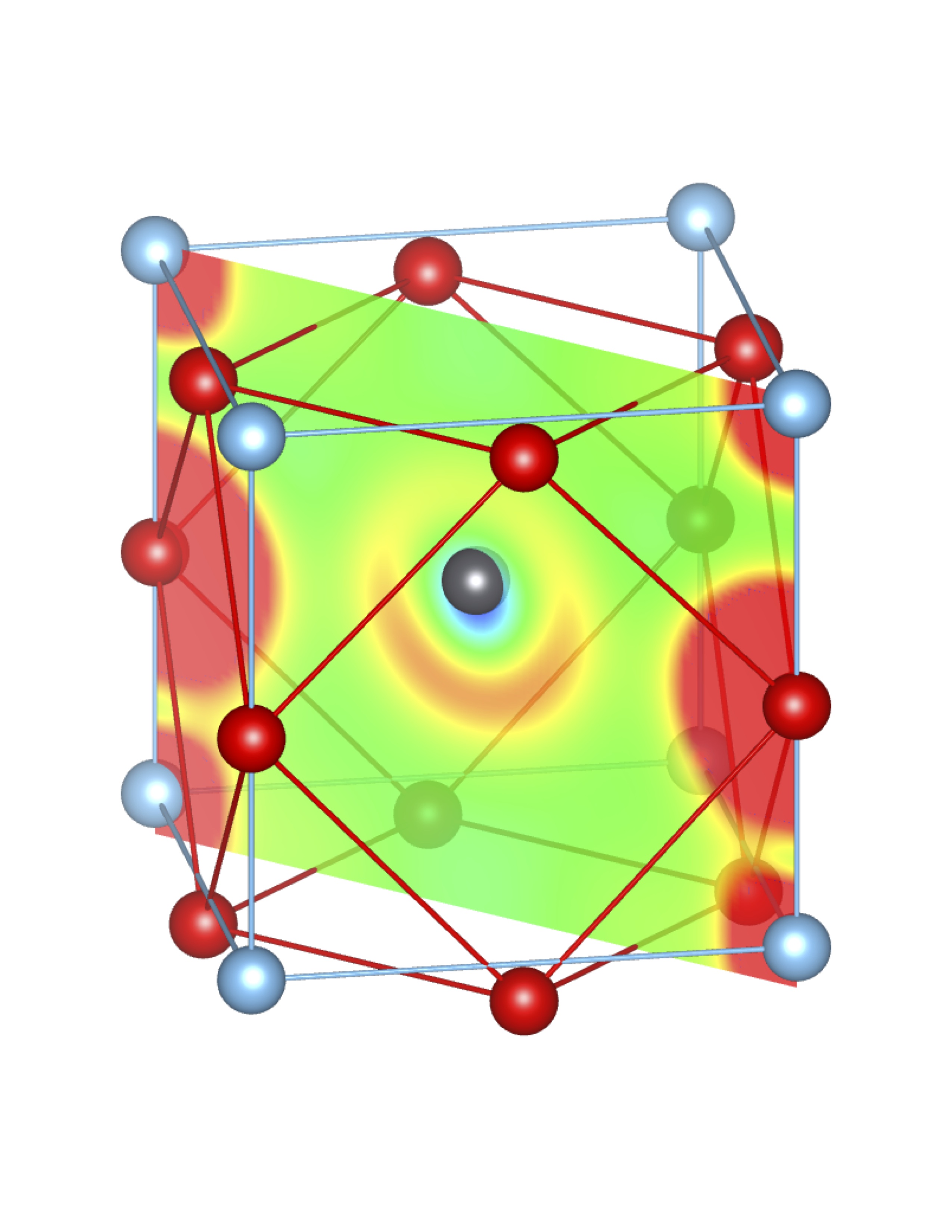 Total Charge (Q) = 0.35 e Total Charge (Q) = 0.35 e |
 Total Charge (Q) = 0.33 e Total Charge (Q) = 0.33 e |
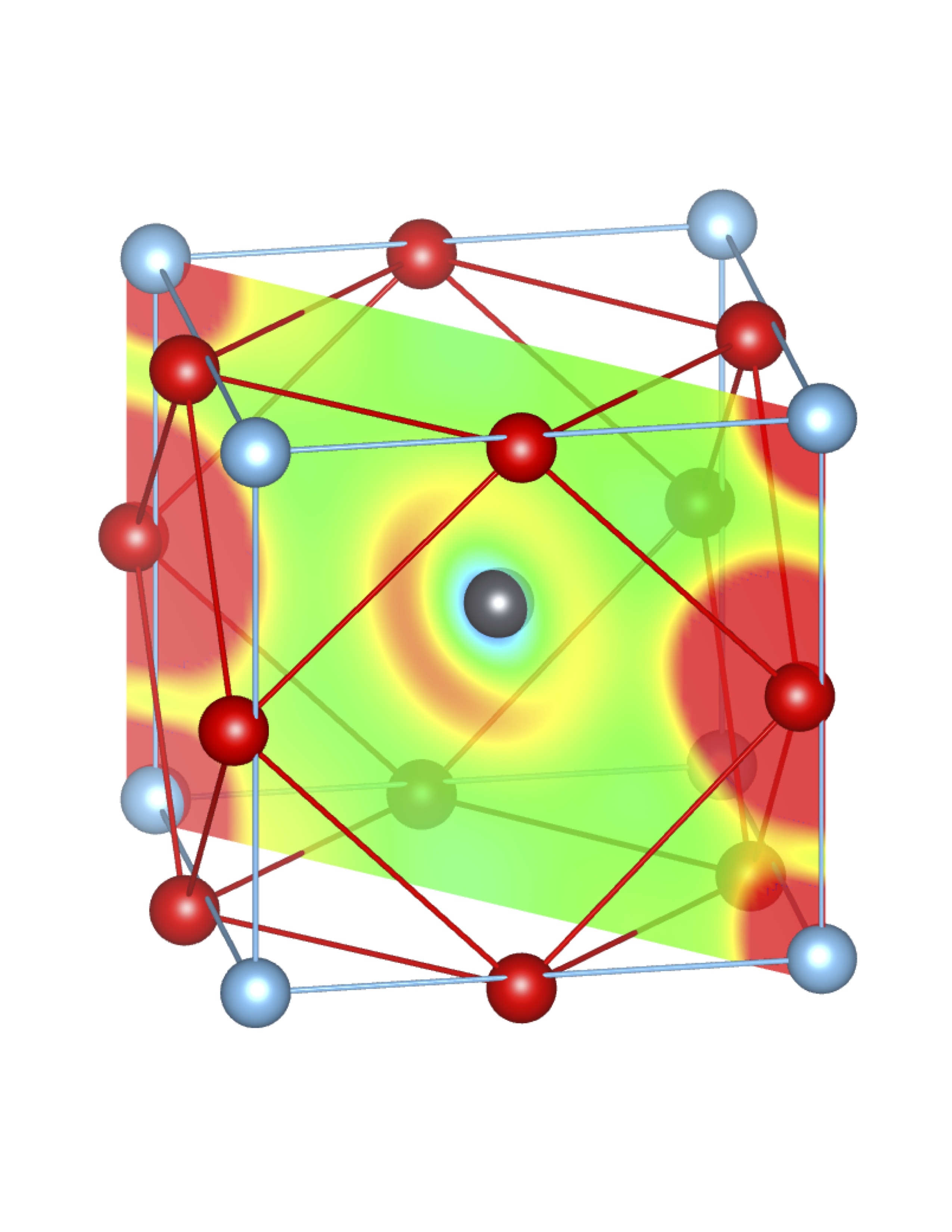 Total Charge (Q) = 0.34 e Total Charge (Q) = 0.34 e |
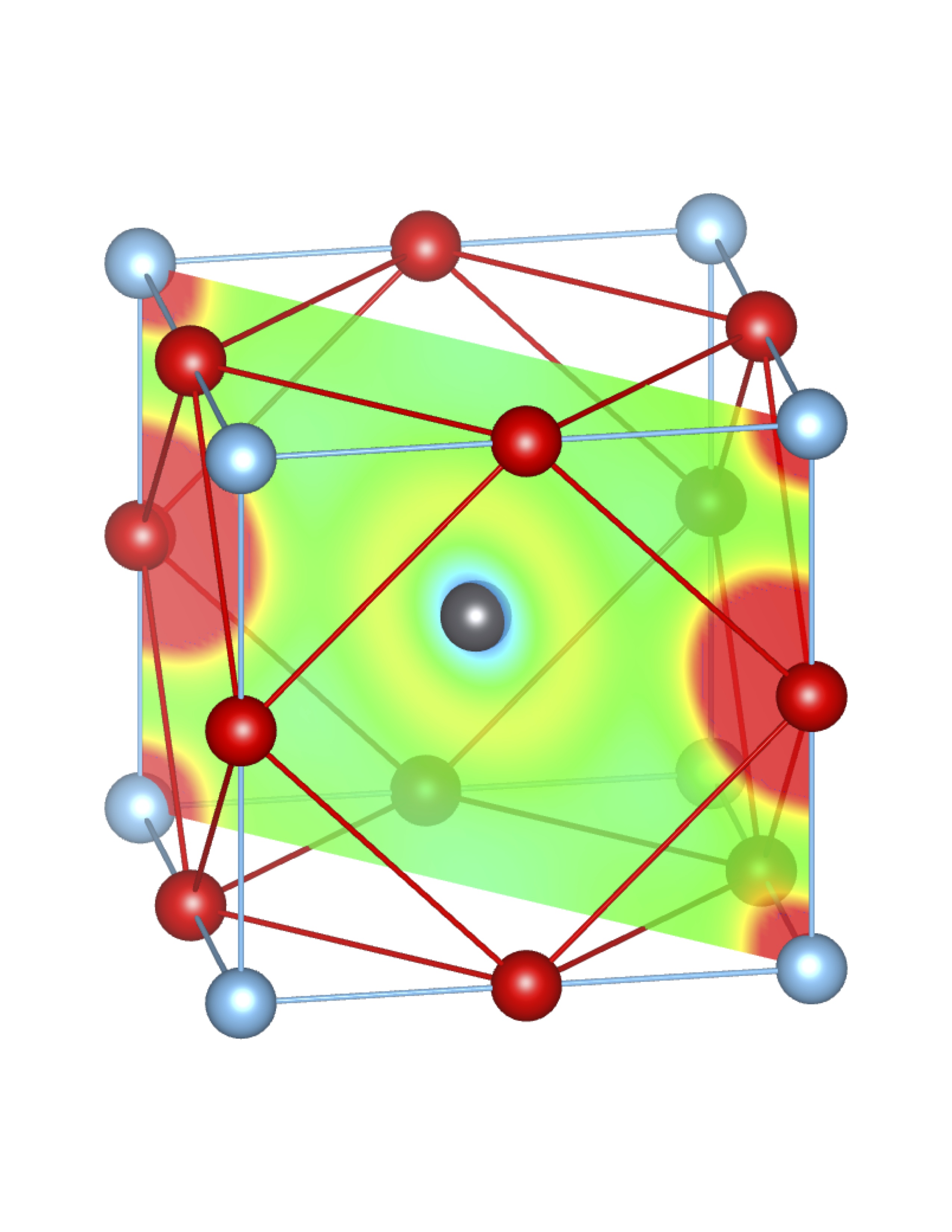 Total Charge (Q) = 0.25 e Total Charge (Q) = 0.25 e |
|
Fig-3: A2+ ion centered unitcells of various polymorphs of SnTiO3 and PbTiO3, presenting the cross section of charge density of lone pairs in [1,1,0] plane.